



29 May 2023 [142] Simple clinical pharmacology can improve prescribing
 Case vignette 1: For painful shingles (H. zoster), a 69 year old took pregabalin 300mg/d and immediate (IR) and slow release (SR) morphine (total 30mg/d). When he developed new “cognitive impairment,” a neurologist recommended he reduce or stop both sedative drugs. Now taking pregabalin at 150mg/d and morphine at only 10mg/d (as IR), he has sweats and mild chills. His “cognitive impairment” is resolved, and thinking his new symptoms may be from opioid withdrawal, your patient telephones for advice. If this is withdrawal, he prefers to “get it over with.” Will knowing the drug half-lives help you respond appropriately?
Case vignette 1: For painful shingles (H. zoster), a 69 year old took pregabalin 300mg/d and immediate (IR) and slow release (SR) morphine (total 30mg/d). When he developed new “cognitive impairment,” a neurologist recommended he reduce or stop both sedative drugs. Now taking pregabalin at 150mg/d and morphine at only 10mg/d (as IR), he has sweats and mild chills. His “cognitive impairment” is resolved, and thinking his new symptoms may be from opioid withdrawal, your patient telephones for advice. If this is withdrawal, he prefers to “get it over with.” Will knowing the drug half-lives help you respond appropriately?
Understanding clinical pharmacology may be under-appreciated in health care. The explosion of new drug categories and curriculum changes have eroded many clinicians’ knowledge of drugs, and no consensus exists on what is most important to learn.1-3 Recent interviews with Canadian prescribers identified limited drug knowledge as one barrier to safe prescribing and deprescribing.4 Reliance on guidelines can camouflage “pharmacoignorance” and even clinicians with specific pharmacological training do not always apply their knowledge effectively to medication reviews.5 Pharmacokinetic (PK) concepts may seem abstruse; but whether we think about them or not, they influence our daily clinical practice. This Therapeutics Letter explores PK principles that can help us prescribe efficiently and avoid common problems.
How can PK parameters help with prescribing?
Pharmacokinetics refers to how drugs enter the body, are distributed and metabolized, and how they finally leave. Drug distribution within the body can include “compartments” with different PK characteristics: blood, interstitial spaces, brain, liver, kidney, lungs, heart, muscle, fat, skin, prostate and eyes. Changes from infancy to old age, genetic or environmental factors, and medical conditions affecting absorption (celiac, short gut) or excretion (kidney or severe liver disease) are sometimes important. Non-lovers of mathematics may find PK intimidating,6 but the simpler concepts can be surprisingly helpful when applied to prescribing, timing of clinical re-assessments, and deprescribing.
Knowing the approximate time when a drug reaches its maximal concentration in blood (Tmax) helps predict onset of effects for drugs that work rapidly (e.g. naproxen, most antibiotics). Knowing the average time for drug concentration to fall by half (T½) may suggest a minimum duration for therapeutic trials. It also suggests how soon to expect any withdrawal symptoms after deprescribing. The Table includes illustrative examples. Therapeutics Letter 134 showed that therapeutic trials of pain drugs are often longer than randomized trials suggest should be necessary.7
Tmax predicts onset of effects
Tmax is the time to reach peak concentration in blood after a drug is administered. In the Figure the blue arrow shows Tmax after a dose of a hypothetical drug. For oral drugs Tmax can depend upon the fed or unfed state and delay in stomach emptying. Drugs given by intravenous (IV) bolus reach Tmax almost instantaneously, and some inhaled drugs behave similarly (e.g. oxygen, nicotine, cannabis). Maximal absorption is also prompt after intramuscular or subcutaneous injections of opioids, naloxone, epinephrine or other drugs given for emergencies.
T½ predicts time to accumulate or dissipate
Half-life (T½) is the mean time for a drug’s concentration to decrease by half after reaching its peak in blood (Figure). T½ can vary substantially; but the mean can be useful to predict the duration of most drug effects. For drugs given intravenously the distribution half-life (T½⍺) is important to anaesthesiologists and emergency personnel (not shown in Figure). Fat-soluble drugs like propofol, fentanyl, or midazolam distribute swiftly from blood into the brain, which receives 20% of resting cardiac output. But these drugs soon redistribute to a much larger mass of body fat outside the brain. Declining drug concentration in the brain determines the rapid offset of effects — even while most of the drug remains elsewhere in the body.
Elimination half-life (T½ elim) is more broadly relevant to clinical decisions. Usually shown as T½, it predicts duration of effects for oral drugs, and for IV drugs after redistribution. The Figure shows how T½ suggests when to expect “steady state” during repeated dosing, when drug elimination balances input. Usually cited as 4 to 5 times the expected half-life, “steady state” is approached by 3 to 4 times T½.
Case vignette 1 – resolution: This man said he preferred to “get it over with” if his symptoms were from withdrawal. Given morphine’s mean T½ of 2 to 4 h, the worst should be over within 24 h. With normal kidneys, pregabalin’s mean T½ is 6 h, so he can stop pregabalin next. He stopped both, without problem. Had he missed either drug, he could have regained “steady state” within ½ to 1 day.
Finding and using Tmax and T½ in practice
Case vignette 2: A patient says “That pill helped my pain fast, but I wish I didn’t have to wait so long to take another.” What if you were in her place?
Tmax and T½ are usually reported as means from PK experiments in small groups. Sometimes the inter-individual range is important and depends (for T½) on genetic polymorphisms (Table). PK values appear in the Clinical Pharmacology sections of online drug monographs, or can be found by a specific search: e.g., “cyclobenzaprine half-life” (mean 18 h). Finding inter-individual ranges may take more work. When a patient says a drug wears off faster or lasts much longer than expected, her individual T½ may differ from the reported mean.
Case vignette 2 – resolution: “listen to the patient – she is telling you the kinetics.” 8 If the drug has a rapid Tmax but a short T½ of 1 to 4 hours, consider more frequent dosing or a formulation that delays absorption.
Remembering drug elimination
Most drugs are cleared by liver metabolism and/or renal excretion. When proportional to blood concentration, this is termed “first order” kinetics. Once a drug is stopped the concentration should decline exponentially (Figure). T½ suggests when concentration will be negligible — although inter-individual variability of T½ and reduced kidney function or severe liver disease can surprise us (Table). Important exceptions include alcohol, phenytoin, or acetaminophen in toxic overdose. When their concentration saturates metabolic pathways, these are cleared at a fixed rate — “zero order” metabolism.
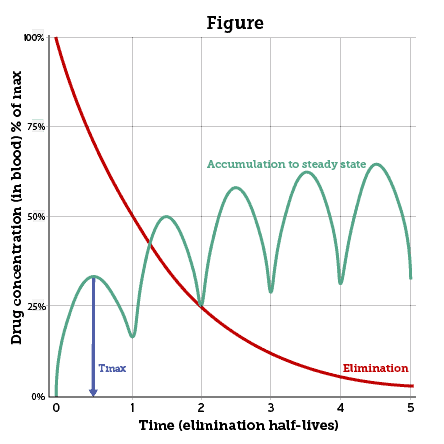
Vertical axis: drug concentration after each dose, as % of maximum attained, absolute values depend on dose, volume of distribution (Vd) and body mass. Horizontal axis: arbitrary units of T½. Blue arrow: Tmax. Green line: “steady state” is approached by 4-5 half-lives. Red line: “first order” exponential decline after final dose (metabolism not saturated, liver/kidney function constant).
Case vignette 3: A 73 year-old with estimated GFR of 12mL/min took gabapentin 300mg tid for painful diabetic neuropathy. By day 10 he felt “too sick to attend my appointment” but was called in and hospitalized. The video shows how predictable pharmacokinetics affected his recovery from acute gabapentin toxicity.
Case vignette 4: A 68 year-old inadvertently took too much phenytoin after her dose per capsule changed. How long after stopping phenytoin will she recover? The video shows how adverse drug effects relate to initial zero order pharmacokinetics.
In a clinical scenario when too much drug puts a patient in danger and the drug has a long T½ or its elimination may be zero order, stop the drug immediately. Wait for the blood concentration to fall, and drug effects will wane.
The Table shows examples for which simple PK understanding is clinically helpful.
‘Long-acting’ formulations
When a rapidly absorbed drug is also eliminated quickly, extending Tmax can prolong its effects. Oral controlled, extended, or slow release (CD/CR/ER/LA/SR/XR) formulations postpone absorption; they do not change T½. Once absorption is complete (typically 12-24 h), drug concentration falls just as it would if the peak occurred earlier. For cancer pain, the smoother concentration profile was a major 1980’s breakthrough for morphine analgesia (T½ 2 to 4 h). Competitors soon followed. For many people with non-cancer pain, predictable tolerance and pharmacologic dependence arising from continuous drug exposure brought significant and sometimes fatal consequences.
Some modified absorption formulations or prodrugs are developed to prolong or avoid patent protection (“evergreening”).9 A recent example is the prodrug lisdexamfetamine (VyvanseTM), which is metabolized to d-amphetamine.10 PK parameters nearly identical to d-amphetamine were demonstrated by academic pharmacologists.11 Transdermal (patch) delivery systems and injectable depot formulations (e.g., antipsychotics, steroids, naltrexone) can also prolong the apparent T½ but like drug metabolism polymorphisms, they are too complex to discuss in this Therapeutics Letter.
Table: Some examples of useful pharmacokinetic parameters and variability
Includes many drugs most often prescribed in BC
Analgesics
| Drug (class) | Main excretion | Tmax (range) | T½ mean (range) | Time to steady state or to dissipate when stopped mean (range) | Comments and references
(Canadian product monographs unless otherwise cited) |
| Acetaminophen | Liver, kidney | 0.4-1 h | 2-3 h | < 1 d | Analgesic effect should be maximal in keeping with kinetics |
| Ibuprofen | Liver, kidney | 1-2 h | 2 h | < 1 d | As above but anti-inflammatory effect might take longer and adverse GI effects can persist longer |
|
Naproxen Enteric coated |
Liver, kidney |
2-3 h 4.2-4.5 h |
13 h | 2-3 d | Adverse effects on stomach may last much longer |
|
Morphine IR Morphine ER |
Liver, kidney |
IR 0.5-1.5h ER 3-4 h |
2-4 h May appear longer |
< 1 d 1-2 d |
“Long-acting” formulations extend Tmax and allow once/twice daily dosing. T½ may appear longer than for IR, due to delayed ongoing absorption after final dose. |
| Methadone12 | Liver |
2.5-4.4 h (1-6 h) |
25 h (8.5-47 h) |
2-9 d | Product monograph notes large inter-individual variability in T1/2
Liver metabolism by CYP3A4. |
CV drugs
| Drug (class) | Main excretion | Tmax (range) | T½ mean (range) | Time to steady state or to dissipate when stopped mean (range) | Comments and references
(Canadian product monographs unless otherwise cited) |
| ASA13 | GI mucosa, liver | 20 min |
13-19 min (salicylate longer) |
After first dose | Platelet inhibition within 60 min; permanent for 10 day platelet life. Canadian Blood Services allows platelet use 3 days after last dose ASA. |
|
Metoprolol IR Metoprolol SR |
Liver |
1.5-2 h 4-5 h |
3.5 h (1-9 h) |
1 d | CYP2D6. Poor metabolizers have longer T½ (9h), extensive metabolizers shorter (1h). |
| Bisoprolol | Kidney | 2-4 h | 9-12 h | < 5 d with daily dosing | |
| Amiodarone | 3-12 h |
53 d (26-107 d) |
265 d (130-535 d) |
Antiepileptics
| Drug (class) | Main excretion | Tmax (range) | T½ mean (range) | Time to steady state or to dissipate when stopped mean (range) | Comments and references
(Canadian product monographs unless otherwise cited) |
| Gabapentin,14 pregabalin |
Kidney Kidney |
2-3 h 0.9-1.4 h |
5-7 h 6.3 h |
1-2 d 1-2 d |
In acute shingles pain, gabapentin analgesia peaked at 2.5 h, concurrent with Tmax. |
| Phenytoin | Liver | ||||
|
Carbamazepine IR CBZ suspension Carbamazepine CR |
Liver |
2-24 h 2 h not shown |
16-24 h (9-36 h) |
3-5 d | Carbamazepine induces its own metabolism, as well as other drugs, and is influenced by other enzyme inducers. CR formulation absorption may vary by brand and should be >> 2 h. |
|
Lamotrigine |
Liver | 1-4.8 h | 26-33 h | 5 d |
Psychiatric drugs
| Drug (class) | Main excretion | Tmax (range) | T½ mean (range) | Time to steady state or to dissipate when stopped mean (range) | Comments and references
(Canadian product monographs unless otherwise cited) |
|
Lithium IR15 Lithium SR |
Kidney |
1-6 h 4-18 h |
18-36 h
|
4-8 d | T½ extended during long-term therapy and impaired GFR. Effects of Li+ in brain, kidney, thyroid may last longer than measured half-life. |
| Quetiapine IR
Quetiapine XL Norquetiapine (active metabolite) |
Liver |
< 2h 6 h |
6-7 h 6-7 h 12 h |
1 d 1-2 d 2 d |
Quetiapine is demethylated in the liver to norquetiapine, an active metabolite that is a potent muscarinic antagonist (anticholinergic) whereas quetiapine is not. |
| Amitriptyline16
Nortriptyline (active metabolite) |
Liver |
4-8 h 7-9 h |
(10-28 h) (16->90 h) |
2-6 d 3-20 d |
Amitriptyine is demethylated in the liver to nortriptyline, an active metabolite with longer T½ but otherwise similar pharmacological properties. Poor metabolizers for CYP2D6 have longer T½. |
| Trazodone | Liver | 1-2 h | 7-8h | 1-2 d | |
| Sertraline | Liver | 6-8 h | 24 h | 4-5 d | |
| Bupropion
Bupropion XL 3 active metabolites |
Liver, kidney |
< 2 h < 3 h
|
14 h (8-24) 20-37 h |
4-5 d 4-8 d |
Roles of 3 active metabolites are unknown but may be significant and could prolong T½ and steady state. Polar active metabolites may be affected by reduced kidney function. |
| Venlafaxine ER
Desmethylvenlafaxine (active metabolite) |
Liver, kidney |
6-8 h 6-8 h |
5 (3-7) h | < 3 d | Effective T½ is governed by slower absorption from ER formulation. Active metabolite desmethylvenlafaxine is an example of “evergreening” when re-marketed as brand name Pristiq. |
| Fluoxetine
Norfluoxetine (active metabolite) |
Liver | 6-8 h |
4-6 d 4-16 d |
1-2 months | Fluoxetine is demethylated to the active metabolite norfluoxetine, that accumulates due to > T½ than fluoxetine. This also protects against withdrawal due to slow elimination. |
| Zopiclone | Liver | < 2 h | 5 h (4-7) | First night | T½ somewhat longer in elderly and severe liver disease. |
| Clonazepam | Liver | 1-4 h | 30-40 h | 5-8 d | Despite long T½ dosing is conventionally divided to minimize peak sedative effects. |
Antibiotics
| Drug (class) | Main excretion | Tmax (range) | T½ mean (range) | Time to steady state or to dissipate when stopped mean (range) | Comments and references
(Canadian product monographs unless otherwise cited) |
|
Penicillin G or V Benzathine |
Kidney |
< 1 h Slow |
< 1 h Delayed by absorption |
First dose | Penicillin is excreted almost immediately by renal tubular secretion unless GFR is nearly zero. Benzathine injection acts as a slowly released depot to deliver low concentations for up to weeks. |
| Amoxicillin | Kidney | 1-2 h | 1 h | First dose | As above |
| Ceftriaxone (IV) | Liver, kidney | immediate | 8 h | ||
| Azithromycin (oral) | Liver | 2 h | 68 h | 12-14 d | Mostly excreted unchanged in bile. Long T½ relates to tissue storage and slow release. |
| Metronidazole | Kidney | 1-2 h | 6-8 h | 1-2 d | Monograph recommends abstinence from alcohol for at least 1 day after completing use to avoid alcohol reaction. |
Miscellaneous others
| Drug (class) | Main excretion | Tmax (range) | T½ mean (range) | Time to steady state or to dissipate when stopped mean (range) | Comments and references
(Canadian product monographs unless otherwise cited) |
|
Pantoprazole Mg Pantoprazole DR |
Kidney |
2-6 h 2-3 h |
1 h 1 h |
1 d 1 d |
The 24 h action of proton pump inhibitors is to to irreversible covalent binding to proton pumps in the gastric mucosa. |
| Donepezil17 | Liver, kidney | 3-4 h | 3 days | 2 weeks | Benefits are of questionable clinical relevance but adverse effects potentially significant. |
|
Tamsulosin18 Tamsulosin CR |
Liver |
1 h 4-6 h
|
12 h Reported as 5-7 h |
2-3 d | Controlled release formulations extend apparent T½ to 12-15 h. Original cited estimate of Tmax and T½ for the regular compound (not CR) is from 4 men. |
| Cyclobenzaprine19,20 | Liver | 4-5 h | 1-3 d | 4-15 d | Cyclobenzaprine is structurally related to tricyclic antidepressants such as amitriptyline. It shares most of their pharmacological properties. |
| Ipilimumab (IV) | Proteolytic enzymes | Immediate | 15 (7-22) | 2 months after max 4 doses | Immunological effects may not be concentration-dependent and may persist indefinitely with no apparent relation to pharmacokinetics. |
Abbreviations: T1/2, T1/2 elim: elimination half-life; T½α: redistribution half-life for IV drugs; Tmax: time to peak blood concentration after a dose; h: hours; d: days.
Notes:
- Time to steady state or to disipate is based on 4-5 half-lives (mean) and normal liver and kidney function.
- Drugs were selected from amongst the most often prescribed drugs in BC or because they exemplify important or unusual pharmacokinetics. Information is generally from recent Canadian product monographs except where a specific reference is shown.
- This Table can be modified online. If you think of an instructive PK example that may be of general interest, you are welcome to submit a comment and supporting reference in response to this issue of the Therapeutics Letter. Please explain why you think another drug should be added, or explain if you identify a non-trivial error in Table, and supply your reference.
Conclusions
- Knowing Tmax can suggest when to assess symptomatic effects of a drug (good or bad).
- Allowing 4 to 5 half-lives predicts steady state effects of drugs taken for symptoms. When a drug is stopped, expect effects to dissipate or potential withdrawal symptoms to emerge after a similar interval.
- Some half-lives reported as means have significant inter-individual ranges. Patients who report shorter or longer duration of effects than expected may have different elimination kinetics.
- “Steady state” seldom applies in sick people. Acute decline in kidney function or saturated liver metabolism can cause dangerous toxicity: e.g., K+, lithium, gabapentin, pregabalin, acetaminophen, phenytoin, alcohol.
 The Therapeutics Letter is a member of the International Society of Drug Bulletins (ISDB), a world-wide network of independent drug bulletins that aims to promote international exchange of quality information on drugs and therapeutics.
The Therapeutics Letter is a member of the International Society of Drug Bulletins (ISDB), a world-wide network of independent drug bulletins that aims to promote international exchange of quality information on drugs and therapeutics.References
- Young H. Lack of pharmacological training causes overuse and misuse of drugs. CMAJ Canadian Medical Association Journal 2008; 178(3):276. DOI: 10.1503/cmaj.071819
- Brinkman DJ, Tichelaar J, Graaff S, et al. Do final-year medical students have sufficient prescribing competencies? A systematic literature review. British Journal Clinical Pharmacology 2018; 84(4):615-635. DOI: 10.1111/bcp.13491
- Keijsers CJ, Ross S. A pharmacological approach to education. British Journal Clinical Pharmacology 2015; 80(3):329-30. DOI: 10.1111/bcp.12700
- Farrell B, Galley E, Jeffs L, et al. “Kind of blurry”: Deciphering clues to prevent, investigate and manage prescribing cascades. PLoS ONE 2022; 17(8):e0272418. DOI: 10.1371/journal.pone.0272418
- Therapeutics Initiative. Does medication review improve health? Therapeutics Letter 104. January-February 2017;
https://ti.ubc.ca/letter104 - Buxton ILO. Pharmacokinetics: The dynamics of drug absorption, distribution, metabolism, and elimination. Goodman & Gilman’s The Pharmacological Basis of Therapeutics, Chapter 2. 2023 McGraw Hill.
- Therapeutics Initiative. Finding the lowest effective dose for non-opioid analgesics. Therapeutics Letter 134. November-December 2021; https://ti.ubc.ca/letter134
- Adapted from a celebrated aphorism attributed to Sir William Osler. https://thedoctorweighsin.com/listen-to-your-patient-hes-telling-you-the-diagnosis/
- Dickson S. Effect of evergreened reformulations on medicaid expenditures and patient access from 2008 to 2016. Journal of Managed Care & Specialty Pharmacy 2019; 25(7):780-792. DOI: 10.18553/jmcp.2019.18366
- KTakeda Canada Inc. Vyvanse product monograph (revised 2020). https://pdf.hres.ca/dpd_pm/00057141.pdf
- Dolder PC, Strajhar P, Vizeli P, et al. Pharmacokinetics and pharmacodynamics of lisdexamfetamine compared with d-amphetamine in healthy subjects. Frontiers in Pharmacology 2017; 8:617. DOI: 10.3389/fphar.2017.00617
- Ferrari A, Coccia CPR, et al. Methadone-metabolism, pharmacokinetics, and interactions. Pharmacological Research 2004; 40:551-559. DOI: 10.1016/j.phrs.2004.05.002
- Awtry EH, Loscalzo J. Aspirin. Circulation 2000; 101:1206-1218. DOI: 10.1161/01.cir.101.10.1206
- Berry JD, Petersen KL. A single dose of gabapentin reduces acute pain and allodynia in patients with herpes zoster. Neurology 2005; 65(3):444-7. DOI: 10.1212/01.wnl.0000168259.94991.8a
- Couffignal C, Chevillard L, El Balkhi S, et al. The Pharmacokinetics of Lithium. In Malhi GS, Masson M, Bellivier F, Editors. The Science and Practice of Lithium Therapy. Springer 2017. Chapter 2, pp. 25-54.
- Gupta, S.K., Shah, J.C. and Hwang, S.S. Pharmacokinetic and pharmacodynamic characterization of OROS® and immediate-release amitriptyline. British Journal of Clinical Pharmacology 1999; 48:71-78. DOI: 10.1046/j.1365-2125.1999.00973.x
- Therapeutics Initiative. Drugs for Alzheimer’s disease. Therapeutics Letter 56. April-August 2005. https://www.ti.ubc.ca/2005/08/31/drugs-for-alzheimers-disease/
- Absorption, metabolism and excretion of tamsulosin hydrochloride in man. Xenobiotica 1996; 26(6):637-645. DOI: 10.3109/00498259609046739
- Tatiane Maria de Lima Souza Brioschi, Simone Grigoleto Schramm, Eunice Kazue Kano, Eunice Emiko Mori Koono, Ting Hui Ching, Cristina Helena dos Reis Serra, Valentina Porta. Pharmacokinetics and Bioequivalence Evaluation of Cyclobenzaprine Tablets. BioMed Research International 2013; vol. 2013, Article ID 281392. DOI: 10.1155/2013/281392
- Therapeutics Initiative. Is cyclobenzaprine useful for pain? Therapeutics Letter 105. March-April 2017. https://www.ti.ubc.ca/2017/07/24/105-cyclobenzaprine/
No Comments